plastimatch¶
Synopsis¶
plastimatch command [options]
Description¶
The plastimatch executable is used for a variety of operations on either 2D or 3D images, including image registration, warping, resampling, and file format conversion. The form of the options depends upon the command given. The list of possible commands can be seen by simply typing “plastimatch” without any additional command line arguments:
$ plastimatch
plastimatch version 1.10.0
Usage: plastimatch command [options]
Commands:
add adjust average bbox boundary
crop compare compose convert dice
diff dmap dose drr dvh
fdk fill filter gamma header
intersect jacobian lm-warp mabs mask
maximum ml-convert multiply probe register
resample scale segment sift stats
synth synth-vf threshold thumbnail union
warp wed xf-convert xf-invert
For detailed usage of a specific command, type:
plastimatch command
plastimatch add¶
The add command is used to add one or more images together and create an output image. The contributions of the input images can be weighted with a weight vector.
The command line usage is given as follows:
Usage: plastimatch add [options] input_file [input_file ...]
Options:
--average produce an output file which is the average of the
input files (if no weights are specified), or
multiply the weights by 1/n
--output <arg> output image
--weight <arg> specify a vector of weights; the images are
multiplied by the weight prior to adding their
values
Examples¶
To add together files 01.mha, 02.mha and 03.mha, and save the result in the file output.mha, you can run the following command:
plastimatch add --output output.mha 01.mha 02.mha 03.mha
If you wanted output.mha to be 2 * 01.mha + 0.5 * 02.mha + 0.1 * 03.mha, then you should do this:
plastimatch add \
--output output.mha \
--weight "2 0.5 0.1" \
01.mha 02.mha 03.mha
plastimatch adjust¶
The adjust command is used to adjust the intensity values within an image. The adjustment operations available are truncation, linear scaling, histogram matching as well as global and local linear matching.
The command line usage is given as follows:
Usage: plastimatch adjust [options]
Options:
-h, --help display this help message
--hist-levels <arg> number of histogram bins for histogram
matching, default is 1024
--hist-match <arg> reference image for histogram matching
--hist-points <arg> number of match points for histogram matching,
default is 10
--hist-threshold threshold at mean intensity (simple background
exclusion) for histogram matching
--input <arg> input directory or filename
--input-mask <arg> input image mask, only affects --linear-match
and --local-match
--linear <arg> shift and scale image intensities, provide a
string with "<shift> <scale>"
--linear-match <arg> reference image for linear matching with mean
and std
--local-match <arg> reference image for patch-wise shift and
scale. You must specify the --patch-size
--local-blending-off no trilinear interpolation of shifts and
scales
--local-scale-out <arg> filename to store pixel-wise scales
--local-shift-out <arg> filename to store pixel-wise shifts
--output <arg> output image
--patch-size <arg> patch size for local matching; provide 1 "n"
or 3 values "nx ny nz"
--pw-linear <arg> a string that forms a piecewise linear map
from input values to output values, of the
form "in1,out1,in2,out2,..."
--ref-mask <arg> reference image mask, only affects
--linear-match and --local-match
--version display the program version
The adjust command can be used to make a piecewise linear adjustment of the image intensities. The –pw-linear option is used to create the mapping from input intensities to output intensities. The input intensities in the curve must increase from left to right in the string, but output intensities are arbitrary. Input intensities below the first pair or after the last pair are transformed by extrapolating the curve out to infinity with a slope of +1. A different slope may be specified out to positive or negative infinity by specifying the special input values of -inf and +inf. In this case, the second number in the pair is the slope of the curve, not the output intensity. You can do a simplified linear transformation of gray levels with the –linear option. For this, you need to provide a string with “<shift> <scale>”.
In addition, you can adjust the image intensities based on a reference image. With –linear-match, a linear transformation (shift and scale) is determined from mean and standard deviation of pixel values in reference and input image. If the input image feaures local intensity inconsistencies, you can choose a patch-based intensity correction using the –local-match option. Similar to –linear-match, shift and scale are computed patch-wise from mean and standard deviation. For both options, you can provide masks that specify the regions taken into account. Finally, choose –hist-match to perform histogram matching.
You can only choose one of –linear, –pw-linear, –linear-match, –local-match and –hist-match. Beware that a reference filename has to be added for matching options.
Examples¶
The following command will add 100 to all voxels in the image:
plastimatch adjust \
--input infile.nrrd \
--output outfile.nrrd \
--pw-linear "0,100"
The following command does the same thing, but with explicit specification of the slope in the extrapolation area:
plastimatch adjust \
--input infile.nrrd \
--output outfile.nrrd \
--pw-linear "-inf,1,0,100,inf,1"
The following command truncates the inputs to the range of [-1000,+1000]:
plastimatch adjust \
--input infile.nrrd \
--output outfile.nrrd \
--pw-linear "-inf,0,-1000,-1000,+1000,+1000,inf,0"
The following command scales and then shifts all voxel values by 2.5 and +1000, respectively. (Use either comma or space to separate the values):
plastimatch adjust \
--input infile.nrrd \
--output outfile.nrrd \
--linear "1000,2.5"
The following command matches the histogram of infile.nrrd to be similar to that of reference.nrrd:
plastimatch adjust \
--input infile.nrrd \
--output outfile.nrrd \
--hist-match reference.nrrd \
--hist-levels 1000 --hist-points 12
The following command matches mean and standard deviation of intensities in the input image to equal those of the reference image:
plastimatch adjust \
--input infile.nrrd \
--output outfile.nrrd \
--linear-match reference.nrrd
The following command also matches mean and standard deviation, but calculates statistics only from inside the mask regions (note that masks are only used for statistic calculations, you would need to use plastimatch mask to reset outside values):
plastimatch adjust \
--input infile.nrrd \
--output outfile.nrrd \
--linear-match reference.nrrd \
--input-mask inmask.nrrd --ref-mask refmask.nrrd
Finally, you can apply patch-wise local intensity adjustment using the following command:
plastimatch adjust \
--input infile.nrrd \
--output outfile.nrrd \
--local-match reference.nrrd \
--patch-size "20 20 10"
The –local-match option requires the input and reference to be spatially aligned. In order to reduce the influence of background pixels at the border, you can provide foreground masks for both images (if only one mask is given, it as also used for the second image):
plastimatch adjust \
--input infile.nrrd
--output outfile.nrrd \
--local-match reference.nrrd \
--patch-size "20 20 10" \
--input-mask inmask.nrrd \
--ref-mask refmask.nrrd
plastimatch average¶
The average command is used to compute the (weighted) average of multiple input images. It is the same as the plastimatch add command, with the –average option specified. Please refer to plastimatch add for the list of command line arguments.
Example¶
The following command will compute the average of three input images:
plastimatch average \
--output outfile.nrrd \
01.mha 02.mha 03.mha
plastimatch autolabel¶
The autolabel command is an experimental program the uses machine learning to identify the thoracic vertibrae in a CT scan.
The command line usage is given as follows:
Usage: plastimatch autolabel [options]
Options:
-h, --help Display this help message
--input <arg> Input image filename (required)
--network <arg> Input trained network filename (required)
--output <arg> Output csv filename (required)
plastimatch boundary¶
The boundary command takes a binary label image as input, and generates an image of the image boundary as the output. The boundary is defined as the voxels within the label which have neighboring voxels outside the label.
The command line usage is given as follows:
Usage: plastimatch boundary [options] input_file
Required:
--output <arg> filename for output image
plastimatch crop¶
The crop command crops out a rectangular portion of the input file, and saves that portion to an output file. The command line usage is given as follows:
Usage: plastimatch crop [options]
Required:
--input=image_in
--output=image_out
--voxels="x-min x-max y-min y-max z-min z-max" (integers)
The voxels are indexed starting at zero.
In other words, if the size of the image is
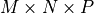 ,
the x values should range between 0 and
,
the x values should range between 0 and 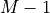 .
.
Example¶
The following command selects the region of size
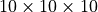 , with the first voxel of the output
image being at location (5,8,12) of the input image:
, with the first voxel of the output
image being at location (5,8,12) of the input image:
plastimatch crop \
--input in.mha \
--output out.mha \
--voxels "5 14 8 17 12 21"
plastimatch compare¶
The compare command compares two files by subtracting one file from the other, and reporting statistics of the difference image. The two input files must have the same geometry (origin, dimensions, and voxel spacing). The command line usage is given as follows:
Usage: plastimatch compare image_in_1 image_in_2
Example¶
The following command subtracts synth_2 from synth_1, and reports the statistics:
$ plastimatch compare synth_1.mha synth_2.mha
MIN -558.201904 AVE 7.769664 MAX 558.680847
MAE 85.100204 MSE 18945.892578
DIF 54872 NUM 54872
The reported statistics are interpreted as follows:
MIN Minimum value of difference image
AVE Average value of difference image
MAX Maximum value of difference image
MAE Mean average value of difference image
MSE Mean squared difference between images
DIF Number of pixels with different intensities
NUM Total number of voxels in the difference image
plastimatch compose¶
The compose command is used to compose two transforms. The command line usage is given as follows:
Usage: plastimatch compose file_1 file_2 outfile
Note: file_1 is applied first, and then file_2.
outfile = file_2 o file_1
x -> x + file_2(x + file_1(x))
The transforms can be of any type, including translation, rigid, affine, itk B-spline, native B-spline, or vector fields. The output file is always a vector field.
There is a further restriction that at least one of the input files must be either a native B-spline or vector field. This restriction is required because that is how the resolution and voxel spacing of the output vector field is chosen.
Example¶
Suppose we want to compose a rigid transform (rigid.tfm) with a vector field (vf.mha), such that the output transform is equivalent to applying the rigid transform first, and the vector field second.
plastimatch compose rigid.tfm vf.mha composed_vf.mha
plastimatch convert¶
The convert command is used to convert files from one format to another format. As part of the conversion process, it can also apply (linear or deformable) geometric transforms to the input images. In fact, convert is just an alias for the warp command.
The command line usage is given as follows:
Usage: plastimatch convert [options]
Options:
--algorithm <arg> algorithm to use for warping, either
"itk" or "native", default is native
--ctatts <arg> ct attributes file (used by dij warper)
--default-value <arg> value to set for pixels with unknown
value, default is 0
--dicom-with-uids <arg> set to false to remove uids from created
dicom filenames, default is true
--dif <arg> dif file (used by dij warper)
--dim <arg> size of output image in voxels "x [y z]"
--direction-cosines <arg>
oriention of x, y, and z axes; Specify
either preset value,
{identity,rotated-{1,2,3},sheared}, or 9
digit matrix string "a b c d e f g h i"
--dose-scale <arg> scale the dose by this value
--fixed <arg> fixed image (match output size to this
image)
--input <arg> input directory or filename; can be an
image, structure set file (cxt or
dicom-rt), dose file (dicom-rt,
monte-carlo or xio), dicom directory, or
xio directory
--input-cxt <arg> input a cxt file
--input-dose-ast <arg> input an astroid dose volume
--input-dose-img <arg> input a dose volume
--input-dose-mc <arg> input an monte carlo volume
--input-dose-xio <arg> input an xio dose volume
--input-prefix <arg> input a directory of structure set
images (one image per file)
--input-ss-img <arg> input a structure set image file
--input-ss-list <arg> input a structure set list file
containing names and colors
--interpolation <arg> interpolation to use when resampling,
either "nn" for nearest neighbors or
"linear" for tri-linear, default is
linear
--metadata <arg> patient metadata (you may use this
option multiple times), option written
as "XXXX,YYYY=string"
--modality <arg> modality metadata: such as {CT, MR, PT},
default is CT
--origin <arg> location of first image voxel in mm "x y
z"
--output-colormap <arg> create a colormap file that can be used
with 3d slicer
--output-cxt <arg> output a cxt-format structure set file
--output-dicom <arg> create a directory containing dicom and
dicom-rt files
--output-dij <arg> create a dij matrix file
--output-dose-img <arg> create a dose image volume
--output-img <arg> output image; can be mha, mhd, nii,
nrrd, or other format supported by ITK
--output-labelmap <arg> create a structure set image with each
voxel labeled as a single structure
--output-pointset <arg> create a pointset file that can be used
with 3d slicer
--output-prefix <arg> create a directory with a separate image
for each structure
--output-prefix-fcsv <arg>
create a directory with a separate fcsv
pointset file for each structure
--output-ss-img <arg> create a structure set image which
allows overlapping structures
--output-ss-list <arg> create a structure set list file
containing names and colors
--output-type <arg> type of output image, one of {uchar,
short, float, ...}
--output-vf <arg> create a vector field from the input xf
--output-xio <arg> create a directory containing xio-format
files
--patient-id <arg> patient id metadata: string
--patient-name <arg> patient name metadata: string
--patient-pos <arg> patient position metadata: one of
{hfs,hfp,ffs,ffp}
--prefix-format <arg> file format of rasterized structures,
either "mha" or "nrrd"
--prune-empty delete empty structures from output
--referenced-ct <arg> dicom directory used to set UIDs and
metadata
--series-description <arg>
series description metadata: string
--simplify-perc <arg> delete <arg> percent of the vertices
from output polylines
--spacing <arg> voxel spacing in mm "x [y z]"
--version display the program version
--xf <arg> input transform used to warp image(s)
--xor-contours overlapping contours should be xor'd
instead of or'd
About referenced-ct¶
The referenced-ct option allows you to (a) create a dicom object which refers to existing objects and (b) copy metadata. The first function, referring to existing objects, uses the following logic to determine how references are made.
If the input objects contain a RTSTRUCT but not an Image (CT), the Frame Of Reference UID (0020,0052), the Series Instance UID (0020,000e) within the RT Referenced Series Sequence (3006,0014), the Referenced SOP Instance UIDs (0008,1155), and the Referenced Frame Of Reference UID (3006,0024) are all set to UIDs of the Image of the referenced-ct.
If the input objects contain a RTDOSE but not a RTSTRUCT, the Referenced Structure Set Sequence (0008,1155) is set to the UID of the RTSTRUCT of the referenced-ct.
If the input objects contain a RTDOSE but not a RTPLAN, the Referenced Structure Set Sequence (0008,1155) is set to the UID of the RTPLAN of the referenced-ct.
For the second function, the following data are copied from the referenced-ct. They are copied from the Image.
Patient Name (0010,0010)
Patient ID (0010,0020)
Examples¶
The first example demonstrates how to convert a DICOM volume to NRRD. The DICOM images that comprise the volume must be stored in a single directory, which for this example is called “dicom-in-dir”. Because the –output-type option was not specified, the output type will be matched to the type of the input DICOM volume. The format of the output file (NRRD) is determined from the filename extension.
plastimatch convert \
--input dicom-in-dir \
--output-img outfile.nrrd
This example further converts the type of the image intensities to float.
plastimatch convert \
--input dicom-in-dir \
--output-img outfile.nrrd \
--output-type float
The next example shows how to resample the output image to a different geometry. The –origin option sets the position of the (center of) the first voxel of the image, the –dim option sets the number of voxels, and the –spacing option sets the distance between voxels. The units for origin and spacing are assumed to be millimeters.
plastimatch convert \
--input dicom-in-dir \
--output-img outfile.nrrd \
--origin "-200 -200 -165" \
--dim "250 250 110" \
--spacing "2 2 2.5"
Generally speaking, it is tedious to manually specify the geometry of the output file. If you want to match the geometry of the output file with an existing file, you can do this using the –fixed option.
plastimatch convert \
--input dicom-in-dir \
--output-img outfile.nrrd \
--fixed reference.nrrd
This next example shows how to convert a DICOM RT structure set file into an image using the –output-ss-img option. Because structures in DICOM RT are polylines, they are rasterized to create the image. The voxels of the output image are 32-bit integers, where the i^th bit of each integer has value one if the voxel lies with in the corresponding structure, and value zero if the voxel lies outside the structure. The structure names are stored in separate file using the –output-ss-list option.
plastimatch convert \
--input structures.dcm \
--output-ss-img outfile.nrrd \
--output-ss-list outfile.txt
In the previous example, the geometry of the output file wasn’t specified. When the geometry of a DICOM RT structure set isn’t specified, it is assumed to match the geometry of the DICOM (CT, MR, etc) image associated with the contours. If the associated DICOM image is in the same directory as the structure set file, it will be found automatically. Otherwise, we have to tell plastimatch where it is located with the –referenced-ct option.
plastimatch convert \
--input structures.dcm \
--output-ss-img outfile.nrrd \
--output-ss-list outfile.txt \
--referenced-ct ../image-directory
plastimatch dice¶
The plastimatch dice compares binary label images using Dice coefficient, Hausdorff distance, or contour mean distance. The input images are treated as boolean, where non-zero values mean that voxel is inside of the structure and zero values mean that the voxel is outside of the structure.
The command line usage is given as follows:
Usage: plastimatch dice [options] reference-image test-image
Options:
--all Compute Dice, Hausdorff, and contour mean
distance (equivalent to --dice --hausdorff
--contour-mean)
--contour-mean Compute contour mean distance
--dice Compute Dice coefficient (default)
--hausdorff Compute Hausdorff distance and average Hausdorff
distance
Example¶
The following command computes all three statistics for mask1.mha and mask2.mha:
plastimatch dice --all mask1.mha mask2.mha
plastimatch diff¶
The plastimatch diff command subtracts one image from another, and saves the output as a new image. The two input files must have the same geometry (origin, dimensions, and voxel spacing).
The command line usage is given as follows:
Usage: plastimatch diff image_in_1 image_in_2 image_out
Example¶
The following command computes file1.nrrd minus file2.nrrd, and saves the result in outfile.nrrd:
plastimatch diff file1.nrrd file2.nrrd outfile.nrrd
plastimatch dmap¶
The plastimatch dmap command takes a binary label image as input, and creates a distance map image as the output. The output image has the same image geometry (origin, dimensions, voxel spacing) as the input image.
The command line usage is given as follows:
Usage: plastimatch dmap [options]
Required:
--input <arg> input directory or filename
--output <arg> output image
Optional:
--algorithm <arg> a string that specifies the algorithm used
for distance map calculation, either
"maurer", "danielsson", or "itk-danielsson"
(default is "danielsson")
--inside-positive voxels inside the structure should be
positive (by default they are negative)
--maximum-distance <arg>
voxels with distances greater than this
number will have the distance truncated to
this number
--squared-distance return the squared distance instead of
distance
Example¶
The following command computes a distance map file dmap.nrrd from a binary labelmap image label.nrrd.:
plastimatch dmap --input label.nrrd --output dmap.nrrd
plastimatch drr¶
A digitally reconstructed radiograph (DRR) is a synthetic radiograph which can be generated from a computed tomography (CT) scan. It is used as a reference image for verifying the correct setup position of a patient prior to radiation treatment.
The plastimatch drr command takes a CT image as input, and generates one or more output images. The output images can be either pgm, pfm, or raw format. The command line usage is:
Usage: plastimatch drr [options] [infile]
Options:
-i, --algorithm <arg> Choose algorithm {exact,uniform}
--autoscale Automatically rescale intensity
--autoscale-range <arg> Range used for autoscale in form "min
max" (default: "0 255")
-z, --detector-size <arg> The physical size of the detector in
format "row col", in mm
-r, --dim <arg> The output resolution in format "row
col" (in mm)
-e, --exponential Do exponential mapping of output values
-y, --gantry-angle <arg> Gantry angle for image source in degrees
-N, --gantry-angle-spacing <arg>
Difference in gantry angle spacing in
degrees
-G, --geometry-only Create geometry files only
-h, --help display this help message
-P, --hu-conversion <arg> Choose HU conversion type
{preprocess,inline,none}
-c, --image-center <arg> The image center in the format "row
col", in pixels
-I, --input <arg> Input file
-s, --intensity-scale <arg> Scaling factor for output image
intensity
-o, --isocenter <arg> Isocenter position "x y z" in DICOM
coordinates (mm)
-n, --nrm <arg> Normal vector of detector in format "x y
z"
-a, --num-angles <arg> Generate this many images at equal
gantry spacing
-O, --output <arg> Prefix for output file(s)
-t, --output-format <arg> Select output format {pgm, pfm, raw}
-S, --raytrace-details <arg>
Create output file with complete ray
trace details
--sad <arg> The SAD (source-axis-distance) in mm
(default: 1000)
--sid <arg> The SID (source-image-distance) in mm
(default: 1500)
-w, --subwindow <arg> Limit DRR output to a subwindow in
format "r1 r2 c1 c2",in pixels
-A, --threading <arg> Threading option {cpu,cuda,opencl}
(default: cpu)
--version display the program version
--vup <arg> The vector pointing from the detector
center to the top row of the detector in
format "x y z"
An input file is required. The drr command can be used in either single image mode or rotational mode. In single image mode, you specify the complete geometry of the x-ray source and imaging panel for a single image. In rotational mode, the imaging geometry is rotated in a circular arc around the isocenter, with a fixed source to axis distance (SAD), and projection images generated at fixed angular intervals.
Examples¶
The following example illustrates the use of single image mode:
plastimatch drr -nrm "1 0 0" \
-vup "0 0 1" \
-g "1000 1500" \
-r "1024 768" \
-z "400 300" \
-c "383.5 511.5" \
-o "0 -20 -50" \
input_file.mha
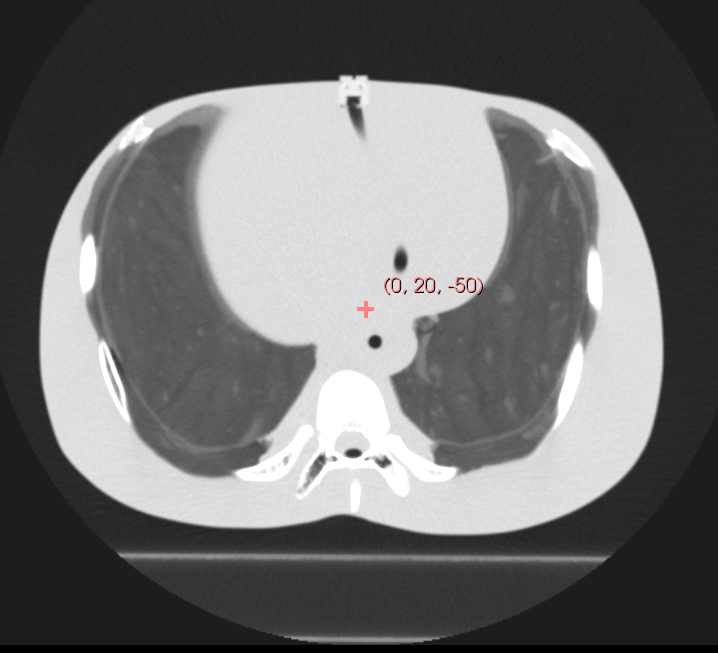
In the above example, the isocenter is chosen to be (0, -20, -50), the location marked on the CT image. The orientation of the projection image is controlled by the nrm and vup options. Using the default values of (1, 0, 0) and (0, 0, 1) yields the DRR shown on the right:
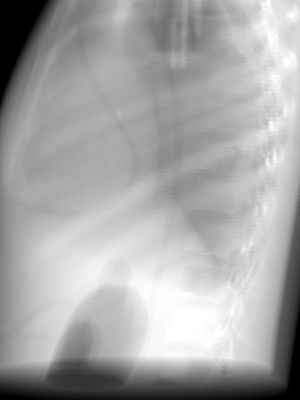
By changing the normal direction (nrm), we can choose different beam direction within an isocentric orbit. For example, an anterior-posterior (AP) DRR is generated with a normal of (0, -1, 0) as shown below:
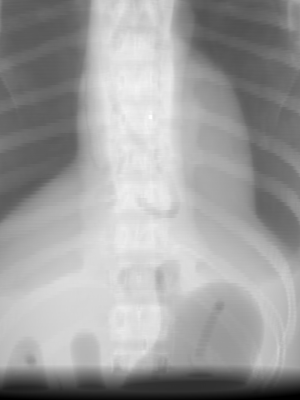
The rotation of the imaging panel is selected using the vup option. The default value of vup is (0, 0, 1), which means that the top of the panel is oriented toward the positive z direction in world coordinates. If we wanted to rotate the panel by 45 degrees counter-clockwise on our AP view, we would set vup to the (1, 0, 1) direction, as shown in the image below. Note that vup doesn’t have to be normalized.

In rotional mode, multiple images are created. The source and imaging panel are assumed to rotate in a circular orbit around the isocenter. The circular orbit is performed around the Z axis, and the images are generated every -N ang degrees of the orbit. This is illustrated using the following example:
plastimatch drr -N 20 \
-a 18 \
-g "1000 1500" \
-r "1024 768" \
-z "400 300" \
-o "0 -20 -50" \
input_file.mha
In the above example, 18 images are generated at a 20 degree interval, as follows:
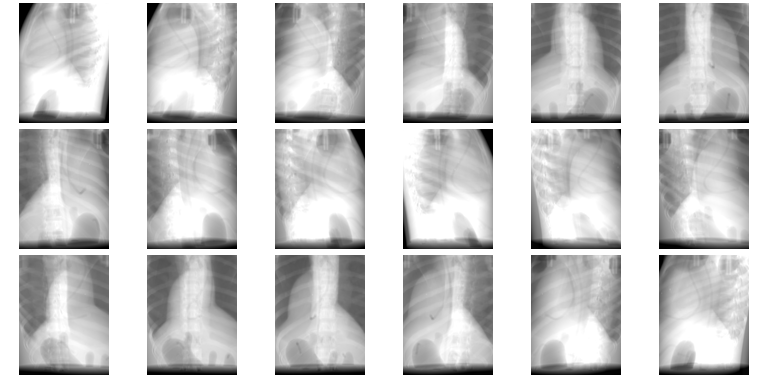
plastimatch dvh¶
The dvh command creates a dose value histogram (DVH) from a given dose image and structure set image. The command line usage is given as follows:
Usage: plastimatch dvh [options]
Options:
--bin-width <arg> specify bin width in the histogram in
units of Gy (default=0.5)
--cumulative create a cumulative DVH (this is the
default)
--differential create a differential DVH instead of a
cumulative DVH
--dose-units <arg> specify units of dose in input file as
either cGy as "cgy" or Gy as "gy"
(default="gy")
-h, --help display this help message
--input-dose <arg> dose image file
--input-ss-img <arg> structure set image file
--input-ss-list <arg> structure set list file containing names
and colors
--normalization <arg> specify histogram values as either voxels
"vox" or percent "pct" (default="pct")
--num-bins <arg> specify number of bins in the histogram
(default=256)
--output-csv <arg> file to save dose volume histogram data in
csv format
--version display the program version
The required inputs are –input-dose, –input-ss-img, –input-ss-list, and –output-csv. The units of the input dose must be either Gy or cGy. DVH bin values will be generated for all structures found in the structure set files. The output will be generated as an ASCII csv-format spreadsheet file, readable by OpenOffice.org or Microsoft Excel.
The default is a differential (standard) histogram, rather than the cumulative DVH which is most common in radiotherapy. To create a cumulative DVH, use the –cumulative option.
The default is to create 256 bins, each with a width of 1 Gy. You can adjust these values using the –num-bins and –bin-width option.
Example¶
To generate a DVH for a single 2 Gy fraction, we might choose 250 bins each of width 1 cGy. If the input dose is already specified in cGy, you would use the following command:
plastimatch dvh \
--input-ss-img structures.mha \
--input-ss-list structures.txt \
--input-dose dose.mha \
--output-csv dvh.csv \
--input-units cgy \
--num-bins 250 \
--bin-width 1
plastimatch fdk¶
The term FDK refers to the authors Feldkamp, Davis, and Kress who wrote the seminal paper “Practical cone-beam algorithm” in 1984. Their paper describes a filtered back-projection reconstruction algorithm for cone-beam geometries. The plastimatch fdk command is an implmenetation of the FDK algorithm. It takes a directory of 2D projection images as input, and generates a single 3D volume as output.
The command line usage is:
Usage: plastimatch fdk [options]
Options:
-x, --detector-offset <arg> The translational offset of the detector
"x0 y0", in pixels
-r, --dim <arg> The output image resolution in voxels
"num (num num)" (default: 256 256 100
-f, --filter <arg> Choice of filter {none,ramp} (default:
ramp)
-X, --flavor <arg> Implementation flavor {0,a,b,c,d}
(default: c)
-h, --help display this help message
-a, --image-range <arg> Use a sub-range of available images
"first ((skip) last)"
-I, --input <arg> Input file
-s, --intensity-scale <arg> Scaling factor for output image
intensity
-O, --output <arg> Prefix for output file(s)
-A, --threading <arg> Threading option {cpu,cuda,opencl}
(default: cpu)
--version display the program version
-z, --volume-size <arg> Physical size of reconstruction volume
"s1 s2 s3", in mm (default: 300 300 150)
The usage of the fdk command is best understood by following along with the tutorials: FDK tutorial (part 1) and FDK tutorial (part 2).
Three different formats of input files are supported. These are:
Pfm format image files with geometry txt files
Raw format image files with geometry txt files
Varian hnd files
The pfm and raw files are similar, in that they store the image as an array of 4-byte little-endian floats. The only difference is that the pfm file has a header which stores the image size, and the raw file does not.
Each pfm or raw image file must have a geometry file in the same directory with the .txt extension. For example, if you want to use image_0000.pfm in a reconstruction, you should supply another file image_0000.txt which contains the geometry. A brief description of the geometry file format is given in Projection matrix file format.
The sequence of files should be stored with the pattern:
XXXXYYYY.ZZZ
where XXXX is a prefix, YYYY is a number, and .ZZZ is the extension of a known type (either .hnd, .pfm, or .raw).
For example the following would be a good directory layout for pfm files:
Files/image_00.pfm
Files/image_00.txt
Files/image_01.pfm
Files/image_01.txt
etc...
The Varian hnd files should be stored in the original layout. For example:
Files/ProjectionInfo.xml
Files/Scan0/Proj_0000.hnd
Files/Scan0/Proj_0001.hnd
etc...
No geometry txt files are needed to reconstruct from Varian hnd format.
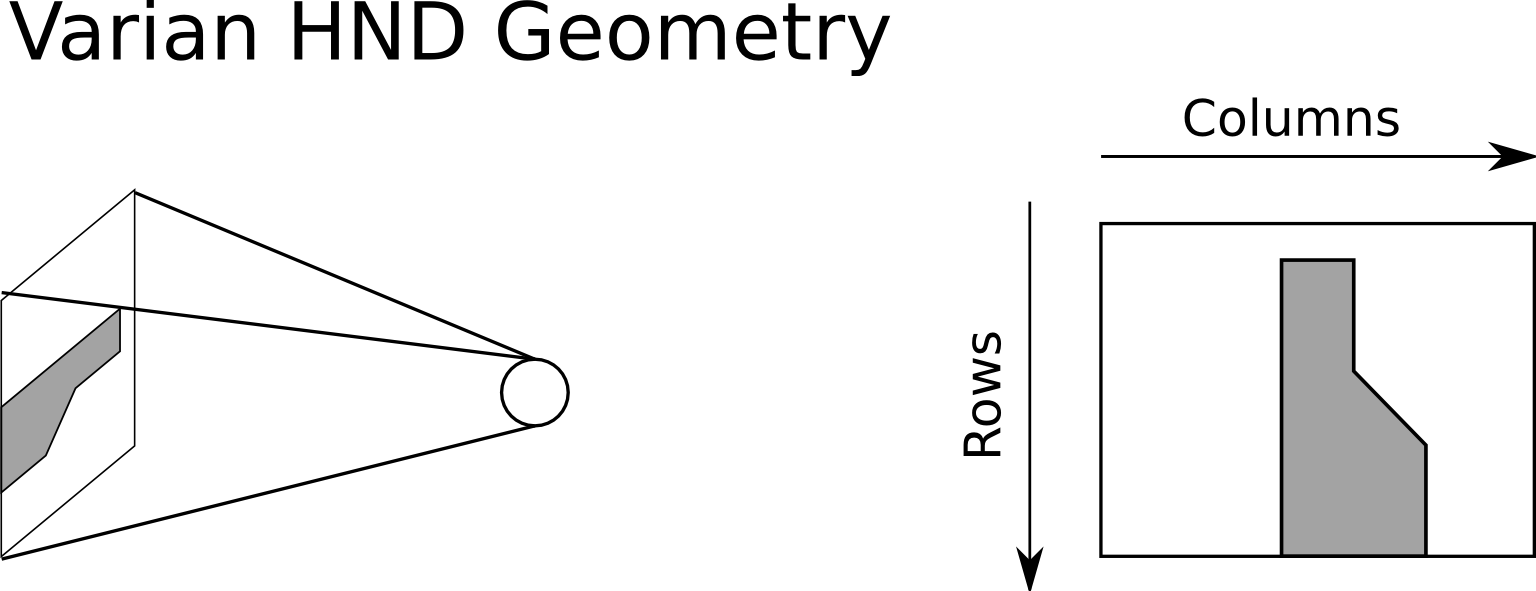
By default, when you generate a DRR, the image is oriented as if the virtual x-ray source were a camera. That means that for a right lateral film, the columns of the image go from inf to sup, and the rows go from ant to post. The Varian OBI system produces HND files, which are oriented differently. For a right lateral film, the columns of the HND images go from ant to post, and the rows go from sup to inf. An illustration of this idea is shown in the figure below.
plastimatch fill¶
The fill command is used to fill an image region with a constant intensity. The region filled is defined by a mask file, with voxels with non-zero intensity in the mask image being filled.
The command line usage is given as follows:
Usage: plastimatch fill [options]
Options:
--input <arg> input directory or filename; can be an image
or dicom directory
--mask <arg> input filename for mask image
--mask-value <arg> value to set for pixels within mask (for
"fill"), or outside of mask (for "mask"
--output <arg> output filename (for image file) or directory
(for dicom)
--output-format <arg> arg should be "dicom" for dicom output
--output-type <arg> type of output image, one of {uchar, short,
float, ...}
Examples¶
Suppose we have a file prostate.nrrd which is zero outside of the prostate, and non-zero inside of the prostate. We can fill the prostate with an intensity of 1000, while leaving non-prostate areas with their original intensity, using the following command.
plastimatch fill \
--input infile.nrrd \
--output outfile.nrrd \
--mask-value 1000 \
--mask prostate.nrrd
plastimatch filter¶
The filter command applies a filter to an input image, and creates a filtered image as its output. The filter can be either built-in, or custom.
The command line usage is given as follows:
Usage: plastimatch filter [options] input_image
Options:
--gabor-k-fib <arg> choose gabor direction at index i within
fibonacci spiral of length n; specified as
"i n" where i and n are integers, and i is
between 0 and n-1
--gauss-width <arg> the width (in mm) of a uniform Gaussian
smoothing filter
--kernel <arg> kernel image filename
--output <arg> output image filename
--output-kernel <arg> output kernel filename
--pattern <arg> filter type: {gabor, gauss, kernel},
default is gauss
The built-in filters supported are “gabor” and “gauss”. For a Gaussian, the width of the Gaussian can be controlled using the –gauss-width option. The Gabor filter is currently limited to automatic selection of filter directions, which are spaced quasi-uniformly on the unit sphere. Custom filters are specified by supplying a kernel file, which is convolved with the image.
Example¶
The following command will generate a filtered image from the first gabor filter within a bank of 10 filters.:
plastimatch filter --pattern gabor Testing/rect-1.mha \
--gabor-k-fib "0 5" --output g-05.mha
plastimatch gamma¶
The gamma command compares two images using the so-called gamma criterion. The gamma criterion specifies that images are similar at a givel location within a reference image if there exists a voxel with similar intensity nearby in the comparison image. Both local gamma and global gamma can be performed using this command.
The command line usage is given as follows:
Usage: plastimatch gamma [options] image_1 image_2
Options:
--analysis-threshold <arg>
Analysis threshold for dose in float (for
example, input 0.1 to apply 10% of the
reference dose). The final threshold dose
(Gy) is calculated by multiplying this
value and a given reference dose (or
maximum dose if not given). (default is
0.1)
--compute-full-region With this option, full gamma map will be
generated over the entire image region
(even for low-dose region). It is
recommended not to use this option to
speed up the computation. It has no
effect on gamma pass-rate.
--dose-tolerance <arg> The scaling coefficient for dose
difference. (e.g. put 0.02 if you want to
apply 2% dose difference criterion)
(default is 0.03)
--dta-tolerance <arg> The distance-to-agreement (DTA) scaling
coefficient in mm (default is 3)
--gamma-max <arg> The maximum value of gamma to compute;
smaller values run faster (default is
2.0)
--inherent-resample <arg>
Spacing value in [mm]. The reference
image itself will be resampled by this
value (Note: resampling compare-image to
ref-image is inherent already). If arg <
0, this option is disabled. (default is
-1.0)
--interp-search With this option, smart interpolation
search will be used in points near the
reference point. This will eliminate the
needs of fine resampling. However, it
will take longer time to compute.
--local-gamma With this option, dose difference is
calculated based on local dose
difference. Otherwise, a given reference
dose will be used, which is called
global-gamma.
--output <arg> Output image
--output-failmap <arg> File path for binary gamma evaluation
result.
--output-text <arg> Text file path for gamma evaluation
result.
--reference-dose <arg> The prescription dose (Gy) used to
compute dose tolerance; if not specified,
then maximum dose in reference volume is
used
--resample-nn With this option, Nearest Neighbor will
be used instead of linear interpolation
in resampling the compare-image to the
reference image. Not recommended for
better results.
Example¶
A gamma image is produced from two input images using the default parameters. This will be a global gamma, using maximum intensity of the reference image as the gamma normalization value.:
plastimatch gamma --output gamma.mha \
reference-image.mha compare-image.mha
plastimatch header¶
The header command is used to display simple properties about the volume, such as the image data type and image geometry.
The command line usage is given as follows:
Usage: plastimatch header [options] input_file [input_file ...]
Options:
-h, --help display this help message
--version display the program version
Example¶
We can display the geometry of any supported file type, such as mha, nrrd, or dicom. We can run the command as follows:
$ plastimatch header input.mha
Type = float
Planes = 1
Origin = -180 -180 -167.75
Size = 512 512 120
Spacing = 0.7031 0.7031 2.5
Direction = 1 0 0 0 1 0 0 0 1
From the header information, we see that the image has 120 slices, and each slice is 512 x 512 pixels. The slice spacing is 2.5 mm, and the in-plane pixel spacing is 0.7031 mm.
plastimatch jacobian¶
The jacobian command computes the Jacobian determinant of a vector field. Either a Jacobian determinant image, or its summary statistics, can be computed.
The command line usage is given as follows:
Usage: plastimatch jacobian [options]
Options:
--input <arg> input directory or filename of image
--output-img <arg> output image; can be mha, mhd, nii, nrrd,
or other format supported by ITK
--output-stats <arg> output stats file; .txt format
Example¶
To create a Jacobian determinant image from a vector field file vf.mha, run the following:
plastimatch jacobian \
--input vf.mha --output-img vf_jac.mha
plastimatch lm-warp¶
The plastimatch lm-warp command performs deformable registration by matching corresponding point landmarks on the fixed and moving images.
The command line usage is given as follows:
Usage: plastimatch lm-warp [options]
Options:
-a, --algorithm <arg> RBF warping algorithm
{tps,gauss,wendland}
-d, --default-value <arg> Value to set for pixels with unknown
value
--dim <arg> Size of output image in voxels "x [y z]"
-F, --fixed <arg> Fixed image (match output size to this
image)
-f, --fixed-landmarks <arg> Input fixed landmarks
-h, --help display this help message
-I, --input-image <arg> Input image to warp
-v, --input-vf <arg> Input vector field (applied prior to
landmark warping)
-m, --moving-landmarks <arg>
Output moving landmarks
-N, --numclusters <arg> Number of clusters of landmarks
--origin <arg> Location of first image voxel in mm "x y
z"
-O, --output-image <arg> Output warped image
-L, --output-landmarks <arg>
Output warped landmarks
-V, --output-vf <arg> Output vector field
-r, --radius <arg> Radius of radial basis function (in mm)
--spacing <arg> Voxel spacing in mm "x [y z]"
-Y, --stiffness <arg> Young modulus (default = 0.0)
--version display the program version
Options “-a”, “-r”, “-Y”, “-d” are set by default to:
-a=gauss Gaussian RBFs with infinite support
-r=50.0 Gaussian width 50 mm
-Y=0.0 No regularization of vector field
-d=-1000 Air
You may want to choose different algorithm:
-a=tps Thin-plate splines (for global registration)
-a=wendland Wendland RBFs with compact support (for
local registration)
In the case of Wendland RBFs “-r” option sets the radius of support.
Regularization of vector field is available for “gauss” and “wendland” algorithms. To regularize the output vector field increase “-Y” to ‘0.1’ and up with increment ‘0.1’.
Example¶
To create a vector field from coresponding landmarks in fixed.fcsv and moving.fcs using Gaussian radial basis functions, do the following:
plastimatch lm-warp \
--output-vf vf.nrrd \
--fixed-landmarks fixed.fcsv --moving-landmarks moving.fcsv
plastimatch mabs¶
The mabs command performs a multi-atlas based segmentation (MABS) operation. The command can operate in one of several training mode, or in segmentation mode.
The command line usage is given as follows:
Usage: plastimatch mabs [options] command_file
Options:
--atlas-selection run just atlas selection
--convert pre-process atlas
--output <arg> output (non-dicom) directory when doing
a segmentation
--output-dicom <arg> output dicom directory when doing a
segmentation
--pre-align pre-process atlas
--segment <arg> use mabs to segment the specified image
or directory
--train perform full training to find the best
registration and segmentation parameters
--train-atlas-selection run just train atlas selection
--train-registration perform limited training to find the
best registration parameters only
Prior to running the mabs command, you must create a configuration file, and you must arrange your training data into the proper directory format. For a complete description of the command file syntax and usage examples, please refer to the Image segmentation (MABS) guidebook and the Image segmentation (MABS) command file reference.
plastimatch mask¶
The mask command is used to fill an image region with a constant intensity. The region filled is defined by a mask file, with voxels with zero intensity in the mask image being filled. Thus, it is the inverse of the fill command.
The command line usage is given as follows:
Usage: plastimatch mask [options]
Options:
--input <arg> input directory or filename; can be an
image or dicom directory
--mask <arg> input filename for mask image
--mask-value <arg> value to set for pixels within mask (for
"fill"), or outside of mask (for "mask"
--output <arg> output filename (for image file) or
directory (for dicom)
--output-format <arg> arg should be "dicom" for dicom output
--output-type <arg> type of output image, one of {uchar, short,
float, ...}
Examples¶
Suppose we have a file called patient.nrrd, which is zero outside of the patient, and non-zero inside the patient. If we want to fill in the area outside of the patient with value -1000, we use the following command.
plastimatch mask \
--input infile.nrrd \
--output outfile.nrrd \
--negate-mask \
--mask-value -1000 \
--mask patient.nrrd
plastimatch ml-convert¶
To be written.
plastimatch probe¶
The plastimatch probe command is used to examine the image intensity or vector field displacement at one or more positions within a volume. The probe positions can be specified in world coordinates (in mm), using the –location option, or as image indices using the –index option. The locations or indices are linearly interpolated if they lie between voxels.
The command line usage is given as follows:
Usage: plastimatch probe [options] file
Options:
-i, --index <arg> List of voxel indices, such as
"i j k;i j k;..."
-l, --location <arg> List of spatial locations, such as
"i j k;i j k;..."
The command will output one line for each probe requested. Each output line includes the following fields.:
PROBE# The probe number, starting with zero
INDEX The (fractional) position of the probe as a voxel index
LOC The position of the probe in world coordinates
VALUE The intensity (for volumes) or displacement
(for vector fields)
Example¶
We use the index option to see an image intensity at coordinate (2,3,4), and the location option to see image intensities at two different locations:
plastimatch probe \
--index "2 3 4" \
--location "0 0 0; 0.5 0.5 0.5" \
infile.nrrd
The output will include three probe results. Each probe shows the probe index, voxel index, voxel location, and intensity.
0: 2.00, 3.00, 4.00; -22.37, -21.05, -19.74; -998.725891
1: 19.00, 19.00, 19.00; 0.00, 0.00, 0.00; -0.000197
2: 19.38, 19.38, 19.38; 0.50, 0.50, 0.50; -9.793450
plastimatch register¶
The plastimatch register command is used to peform linear or deformable registration of two images. The command line usage is given as follows:
Usage: plastimatch register command_file
The command file is an ordinary text file, which contains a single global section and one or more stages sections. The global section begins with a line containing only the string “[GLOBAL]”, and each stage begins with a line containing the string “[STAGE]”.
The global section is used to set input files, output files, and global parameters, while the each stage section defines a sequential stage of processing. For a complete description of the command file syntax, please refer to the Image registration command file reference.
Examples¶
If you want to register image_2.mha to match image_1.mha using B-spline registration, create a command file like this:
# command_file.txt
[GLOBAL]
fixed=image_1.mha
moving=image_2.mha
img_out=warped_2.mha
xform_out=bspline_coefficients.txt
[STAGE]
xform=bspline
impl=plastimatch
threading=openmp
max_its=30
regularization_lambda=0.005
grid_spac=100 100 100
res=4 4 2
Then, run the registration like this:
plastimatch register command_file.txt
The above example only performs a single registration stage. If you want to do multi-stage registration, use multiple [STAGE] sections. Like this:
# command_file.txt
[GLOBAL]
fixed=image_1.mha
moving=image_2.mha
img_out=warped_2.mha
xform_out=bspline_coefficients.txt
[STAGE]
xform=bspline
impl=plastimatch
threading=openmp
max_its=30
regularization_lambda=0.005
grid_spac=100 100 100
res=4 4 2
[STAGE]
max_its=30
grid_spac=80 80 80
res=2 2 1
[STAGE]
max_its=30
grid_spac=60 60 60
res=1 1 1
For more examples, please refer to the Image registration guidebook.
plastimatch resample¶
The resample command can be used to change the geometry of an image.
The command line usage is given as follows:
Usage: plastimatch resample [options]
Options:
--default-value <arg> value to set for pixels with unknown value,
default is 0
--dim <arg> size of output image in voxels "x [y z]"
--direction-cosines <arg>
oriention of x, y, and z axes; Specify either
preset value,
{identity,rotated-{1,2,3},sheared}, or 9 digit
matrix string "a b c d e f g h i"
-F, --fixed <arg> fixed image (match output size to this image)
-h, --help display this help message
--input <arg> input directory or filename; can be an image or
vector field
--interpolation <arg> interpolation type, either "nn" or "linear",
default is linear
--origin <arg> location of first image voxel in mm "x y z"
--output <arg> output image or vector field
--output-type <arg> type of output image, one of {uchar, short,
float, ...}
--spacing <arg> voxel spacing in mm "x [y z]"
--subsample <arg> bin voxels together at integer subsampling rate
"x [y z]"
--version display the program version
Example¶
We can use the –subsample option to bin an integer number of voxels to a single voxel. So for example, if we want to bin a cube of size 3x3x1 voxels to a single voxel, we would do the following.
plastimatch resample \
--input infile.nrrd \
--output outfile.nrrd \
--subsample "3 3 1"
plastimatch scale¶
The scale command scales an image or vector field by multiplying each voxel by a constant value.
The command line usage is given as follows:
Usage: plastimatch scale [options] input_file
Options:
--output <arg> filename for output image or vector field
--weight <arg> scale the input image or vector field by this
value (float)
Example¶
This command creates an output file with image intensity (or voxel length) twice as large as the input values:
plastimatch scale --output output.mha --weight 2.0 input.mha
plastimatch segment¶
The segment command does simple threshold-based semgentation. The command line usage is given as follows:
Usage: plastimatch segment [options]
Options:
-h, --help Display this help message
--input <arg> Input image filename (required)
--lower-threshold <arg> Lower threshold (include voxels
above this value)
--output-dicom <arg> Output dicom directory (for RTSTRUCT)
--output-img <arg> Output image filename
--upper-threshold <arg> Upper threshold (include voxels
below this value)
Example¶
Suppose we have a CT image of a water tank, and we wish to create an image which has ones where there is water, and zeros where there is air. Then we could do this:
plastimatch segment \
--input water.mha \
--output-img water-label.mha \
--lower-threshold -500
If we wanted instead to create a DICOM-RT structure set, we should specify a DICOM image as the input. This will allow plastimatch to create the DICOM-RT with the correct patient name, patient id, and UIDs. The output file will be called “ss.dcm”.
plastimatch segment \
--input water_dicom \
--output-dicom water_dicom \
--lower-threshold -500
plastimatch stats¶
The plastimatch stats command displays a few basic statistics about the image onto the screen.
The command line usage is given as follows:
Usage: plastimatch stats file [file ...]
The input files can be either 2D projection images, 3D volumes, or 3D vector fields.
Example¶
The following command displays statistics for the 3D volume synth_1.mha.
$ plastimatch stats synth_1.mha
MIN -999.915161 AVE -878.686035 MAX 0.000000 NUM 54872
The reported statistics are interpreted as follows:
MIN Minimum intensity in image
AVE Average intensity in image
MAX Maximum intensity in image
NUM Number of voxels in image
Example¶
The following command displays statistics for the 3D vector field vf.mha:
$ plastimatch stats vf.mha
Min: 0.000 -0.119 -0.119
Mean: 13.200 0.593 0.593
Max: 21.250 1.488 1.488
Mean abs: 13.200 0.594 0.594
Energy: MINDIL -6.79 MAXDIL 0.166 MAXSTRAIN 41.576 TOTSTRAIN 70849
Min dilation at: (29 19 19)
Jacobian: MINJAC -6.32835 MAXJAC 1.15443 MINABSJAC 0.360538
Min abs jacobian at: (28 36 36)
Second derivatives: MINSECDER 0 MAXSECDER 388.82 TOTSECDER 669219
INTSECDER 1.524e+06
Max second derivative: (29 36 36)
The rows corresponding to “Min, Mean, Max, and Mean abs” each have three numbers, which correspond to the x, y, and z coordinates. Therefore, they compute these statistics for each vector direction separately.
The remaining statistics are described as follows:
MINDIL Minimum dilation
MAXDIL Maximum dilation
MAXSTRAIN Maximum strain
TOTSTRAIN Total strain
MINJAC Minimum Jacobian
MAXJAC Maximum Jacobian
MINABSJAC Minimum absolute Jacobian
MINSECDER Minimum second derivative
MAXSECDER Maximum second derivative
TOTSECDER Total second derivative
INTSECDER Integral second derivative
plastimatch synth¶
The synth command creates a synthetic image. The following kinds of images can be created, by specifying the appropriate –pattern option. Each of these patterns come with a synthetic structure set and synthetic dose which can be used for testing.
donut – a donut shaped structure
gauss – a Gaussian blur
grid – a 3D grid
lung – a synthetic lung with a tumor
rect – a uniform rectangle within a uniform background
sphere – a uniform sphere within a uniform background
xramp – an image that linearly varies intensities in the x direction
yramp – an image that linearly varies intensities in the y direction
zramp – an image that linearly varies intensities in the z direction
The command line usage is given as follows:
Usage: plastimatch synth [options]
Options:
--background <arg> intensity of background region
--cylinder-center <arg> location of cylinder center in mm "x [y
z]"
--cylinder-radius <arg> size of cylinder in mm "x [y z]"
--dicom-with-uids <arg> set to false to remove uids from created
dicom filenames, default is true
--dim <arg> size of output image in voxels "x [y z]"
--direction-cosines <arg>
oriention of x, y, and z axes; Specify
either preset value,
{identity,rotated-{1,2,3},sheared}, or 9
digit matrix string "a b c d e f g h i"
--donut-center <arg> location of donut center in mm "x [y z]"
--donut-radius <arg> size of donut in mm "x [y z]"
--donut-rings <arg> number of donut rings (2 rings for
traditional donut)
--dose-center <arg> location of dose center in mm "x y z"
--dose-size <arg> dimensions of dose aperture in mm "x [y
z]", or locations of rectangle corners
in mm "x1 x2 y1 y2 z1 z2"
--fixed <arg> fixed image (match output size to this
image)
--foreground <arg> intensity of foreground region
--gabor-k-fib <arg> choose gabor direction at index i within
fibonacci spiral of length n; specified
as "i n" where i and n are integers, and
i is between 0 and n-1
--gauss-center <arg> location of Gaussian center in mm "x [y
z]"
--gauss-std <arg> width of Gaussian in mm "x [y z]"
--grid-pattern <arg> grid pattern spacing in voxels "x [y z]"
--input <arg> input image (add synthetic pattern onto
existing image)
--lung-tumor-pos <arg> position of tumor in mm "z" or "x y z"
--metadata <arg> patient metadata (you may use this
option multiple times)
--noise-mean <arg> mean intensity of gaussian noise
--noise-std <arg> standard deviation of gaussian noise
--origin <arg> location of first image voxel in mm "x y
z"
--output <arg> output filename
--output-dicom <arg> output dicom directory
--output-dose-img <arg> filename for output dose image
--output-ss-img <arg> filename for output structure set image
--output-ss-list <arg> filename for output file containing
structure names
--output-type <arg> data type for output image: {uchar,
short, ushort, ulong, float}, default is
float
--patient-id <arg> patient id metadata: string
--patient-name <arg> patient name metadata: string
--patient-pos <arg> patient position metadata: one of
{hfs,hfp,ffs,ffp}
--pattern <arg> synthetic pattern to create: {cylinder,
donut, dose, gabor, gauss, grid, lung,
noise, rect, sphere, xramp, yramp,
zramp}, default is gauss
--penumbra <arg> width of dose penumbra in mm
--rect-size <arg> width of rectangle in mm "x [y z]", or
locations of rectangle corners in mm "x1
x2 y1 y2 z1 z2"
--spacing <arg> voxel spacing in mm "x [y z]"
--sphere-center <arg> location of sphere center in mm "x y z"
--sphere-radius <arg> radius of sphere in mm "x [y z]"
--volume-size <arg> size of output image in mm "x [y z]"
Examples¶
Create a cubic water phantom 30 x 30 x 40 cm with zero position at the center of the water surface:
plastimatch synth \
--pattern rect \
--output water_tank.mha \
--rect-size "-150 150 0 400 -150 150" \
--origin "-245.5 245.5 -49.5 449.5 -149.5 149.5" \
--spacing "1 1 1" \
--dim "500 500 300"
Create lung phantoms with two different tumor positions, and output to dicom:
plastimatch synth \
--pattern lung \
--output-dicom lung_inhale \
--lung-tumor-pos "0 0 10"
plastimatch synth \
--pattern lung \
--output-dicom lung_exhale \
--lung-tumor-pos "0 0 -10"
plastimatch synth-vf¶
The synth-vf command creates a synthetic vector field. The following kinds of vector fields can be created, by specifying the appropriate option.
gauss – a gaussian warp
radial – a radial expansion or contraction
translation – a uniform translation
zero – a vector field that is zero everywhere
The command line usage is given as follows:
Usage: plastimatch synth-vf [options]
Options:
--dim <arg> size of output image in voxels "x [y z]"
--direction-cosines <arg>
oriention of x, y, and z axes; Specify
either preset value, {identity,
rotated-{1,2,3}, sheared}, or 9 digit
matrix string "a b c d e f g h i"
--fixed <arg> An input image used to set the size of the
output
--gauss-center <arg> location of center of gaussian warp "x [y
z]"
--gauss-mag <arg> displacment magnitude for gaussian warp in
mm "x [y z]"
--gauss-std <arg> width of gaussian std in mm "x [y z]"
--origin <arg> location of first image voxel in mm "x y
z"
--output <arg> output filename
--radial-center <arg> location of center of radial warp "x [y
z]"
--radial-mag <arg> displacement magnitude for radial warp in
mm "x [y z]"
--spacing <arg> voxel spacing in mm "x [y z]"
--volume-size <arg> size of output image in mm "x [y z]"
--xf-gauss gaussian warp
--xf-radial radial expansion (or contraction)
--xf-trans <arg> uniform translation in mm "x y z"
--xf-zero Null transform
plastimatch threshold¶
The threshold command creates a binary labelmap image from an input intensity image.
The command line usage is given as follows:
Usage: plastimatch threshold [options]
Options:
--above <arg> value above which output has value high
--below <arg> value below which output has value high
-h, --help display this help message
--input <arg> input directory or filename
--output <arg> output image
--range <arg> a string that forms a list of threshold ranges of the
form "r1-lo,r1-hi,r2-lo,r2-hi,...", such that voxels
with intensities within any of the ranges
([r1-lo,r1-hi], [r2-lo,r2-hi], ...) have output value
high
--version display the program version
Example¶
The following command creates a binary label image with value 1 when input intensities are between 100 and 200, and value 0 otherwise.:
plastimatch threshold \
--input input_image.nrrd \
--output output_labe.nrrd \
--range "100,200"
plastimatch thumbnail¶
The thumbnail command generates a two-dimensional thumbnail image of an axial slice of the input volume. The output image is not required to correspond exactly to an integer slice number. The location of the output image within the slice is always centered.
The command line usage is given as follows:
Usage: plastimatch thumbnail [options] input-file
Options:
--input file
--output file
--thumbnail-dim size
--thumbnail-spacing size
--slice-loc location
Example¶
We create a two-dimensional image with resolution 10 x 10 pixels, at axial location 0, and of size 20 x 20 mm:
plastimatch thumbnail \
--input in.mha --output out.mha \
--thumbnail-dim 10 \
--thumbnail-spacing 2 \
--slice-loc 0
plastimatch union¶
The union command creates a binary volume which is the logical union of two input images. Voxels in the output image have value one if the voxel is non-zero in either input image, or value zero if the voxel is zero in both input images.
The command line usage is given as follows:
Usage: plastimatch union [options] input_1 input_2
Options:
-h, --help display this help message
--output <arg> filename for output image
--version display the program version
Example¶
The following command creates a volume that is the union of two input images:
plastimatch union \
--output itv.mha \
phase_1.mha phase_2.mha
plastimatch warp¶
The warp command is an alias for convert. Please refer to plastimatch convert for the list of command line parameters.
Examples¶
To warp an image using the B-spline coefficients generated by the plastimatch register command (saved in the file bspline.txt), do the following:
plastimatch warp \
--input infile.nrrd \
--output-img outfile.nrrd \
--xf bspline.txt
In the previous example, the output file geometry was determined by the geometry information in the bspline coefficient file. You can resample to a different geometry using –fixed, or –origin, –dim, and –spacing.
plastimatch warp \
--input infile.nrrd \
--output-img outfile.nrrd \
--xf bspline.txt \
--fixed reference.nrrd
When warping a structure set image, where the integer bits correspond to structure membership, you need to use nearest neighbor interpolation rather than linear interpolation.
plastimatch warp \
--input structures-in.nrrd \
--output-img structures-out.nrrd \
--xf bspline.txt \
--interpolation nn
Sometimes, voxels located outside of the geometry of the input image will be warped into the geometry of the output image. By default, these areas are “filled in” with an intensity of zero. You can choose a different value for these areas using the –default-value option.
plastimatch warp \
--input infile.nrrd \
--output-img outfile.nrrd \
--xf bspline.txt \
--default-value -1000
In addition to images and structures, landmarks exported from 3D Slicer can also be warped.
plastimatch warp \
--input fixed_landmarks.fcsv \
--output-pointset warped_landmarks.fcsv \
--xf bspline.txt
Sometimes, it may be desirable to apply a transform explicitly defined by a vector field instead of using B-spline coefficients. To allow this, the –xf option also accepts vector field volumes. For example, the previous example would become.
plastimatch warp \
--input fixed_landmarks.fcsv \
--output-pointset warped_landmarks.fcsv \
--xf vf.mha
plastimatch xf-convert¶
The xf-convert command converts between transform types. A transform can be either a B-spline transform, or a vector field. There are two different kinds of B-spline transform formats: the plastimatch native format, and the ITK format. In addition to converting the transform type, the xf-convert command can also change the grid-spacing of B-spline transforms.
The command line usage is given as follows:
Usage: plastimatch xf-convert [options]
Options:
--dim <arg> Size of output image in voxels "x [y z]"
--grid-spacing <arg> B-spline grid spacing in mm "x [y z]"
--input <arg> Input xform filename (required)
--nobulk Omit bulk transform for itk_bspline
--origin <arg> Location of first image voxel in mm "x y z"
--output <arg> Output xform filename (required)
--output-type <arg> Type of xform to create (required), choose
from {bspline, itk_bspline, vf}
--spacing <arg> Voxel spacing in mm "x [y z]"
Example¶
We want to convert a B-spline transform into a vector field. If the B-spline transform is in native-format, the vector field geometry is defined by the values found in the transform header.:
plastimatch xf-convert \
--input bspline.txt \
--output vf.mha \
--output-type vf
Likewise, if we want to convert a vector field into a set of B-spline coefficients with a control-point spacing of 30 mm in each direction.
plastimatch xf-convert \
--input vf.mha \
--output bspline.txt \
--output-type bspline \
--grid-spacing 30